
About
About JSpecies
JSpecies is an easy to use, biologist-centric software designed to measure the probability if two genomes belonging to the same species or not.
JSpecies is written in the platform-independent, object-oriented programming language, Java. It can be started using the Java Web Start technology, which automatically downloads and installs the software locally. This ensures the user to always get access to the latest version available. Alternatively, it can be downloaded and installed manually. In order to calculate species relationship two additional tools are needed locally installed: BLAST and MUMmer. Unfortunately, the MUMmer software is not available for any Microsoft operating systems.
-->
The Biology behind
DNA-DNA hybridization (DDH) was firstly implemented in the mid sixties as a genome to genome comparison value trying to find
an objective numerical frontier of what a species could be, and since then it has been recalled to be the gold standard to
genomically circumscribe the basic category in prokaryotic classification. The empirical evidences based on classified species
and their comparisons with DDH values lead to the recommendation that 70% reassociation could be a plausible limit in this
category circumscription. However, the species evaluations made in the following years demonstrated that such limit could not
be taken as an absolute threshold, that due to the large extent of diversity among prokaryotes the circumscription based on
DDH could vary depending on the group in study, and would range between 60 – 70%. Despite some criticisms raised due to the
fact that such delimitation may not help in equalizing eukaryotes as primates and prokaryotic species, microbial taxonomists
agree that it is the best that has been achieved (Rosselló-Móra, 2006).
However, in the age of genomics, there is a need to substitute DDH by means of a method that provides equivalent information,
but is database-based. Among the different approaches developed, the Average Nucleotide Identity (ANI) among the conserved genes
of a pair of genomes can be considered as the first reliable attempt to numerically circumscribe prokaryotic species that may
be equalized to the traditional (DDH) experiments (Konstantinidis and Tiedje, 2005). A more
pragmatic approach to calculate ANI was developed by Goris and colleagues (2007), in where
instead of a previous selection of orthologous genes of a given pair of genomes, the query genome is spliced in 1020 nucleotide
fragments and each of them blasted against the subject genome to calculate the average of nucleotide identity. Both cases lead
to similar results in where the threshold frontier to consider two organisms to belong to the same species could be set at >94%
identity. This value is recalled to substitute the traditional DDH values in the near future.
We have developed the work package JSpecies as a user-friendly, biologist-oriented interface to calculate ANI
(Richter and Rosselló-Móra, 2009). Here you can calculate ANI by using the BLAST as proposed by
Goris and colleagues (2007). Besides, we have implemented an alternative ANI calculation
based on the MUMmer that is designed to compare large DNA stretches and avoids previous manipulation of the sequences,
and renders faster results than BLAST. Both algorithms render near identical results especially in the ANI ranges >90%,
that are those to take into consideration when trying to circumscribe species. In addition, we have implemented the calculation of
the correlation indexes of the tetranucleotide signatures between pairwise genomic comparisons. This parameter is calculated by an
alignment-free analysis and due to the speed of computation allows larger set of genome comparisons.
The decision of enclosing two or more strains in one species should be taken after a thorough evaluation of the genomic and phenotypic
parameters that altogether indicate that the strains share a global coherence that makes them to deserve an independent specific
status (Tindall et al., 2009). The traditional DDH values that recommended the species borders to be set at 60 – 70%, can be
equalized to the ANI range 95 – 96%. However, these values cannot be taken as absolute boundaries unless they agree with the
whole phenotypic and phylogenetic picture.
Reference:
Rosselló-Móra R (2006) DNA-DNA reassociation methods applied to microbial taxonomy and their critical evaluation. in Molecular Identification, Systematics, and Population Structure of Prokaryotes, ed Stackebrandt E (Springer-Verlag, Berlin), pp 23-50
Konstantinidis K, & Tiedje J M (2005) Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA 102, 2567-2592
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P & Tiedje JM (2007) DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 57(Pt 1):81-91
Richter M, & Rosselló-Móra R (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA in press
Tindall BJ, Rosselló-Móra R., Busse H-J, Ludwig W & Kämpfer P (2009) Notes on the characterization of prokaryote strains for taxonomic purposes. Int J Syst Evol Microbiol. [Epub ahead of print]
The Bioinformatics behind
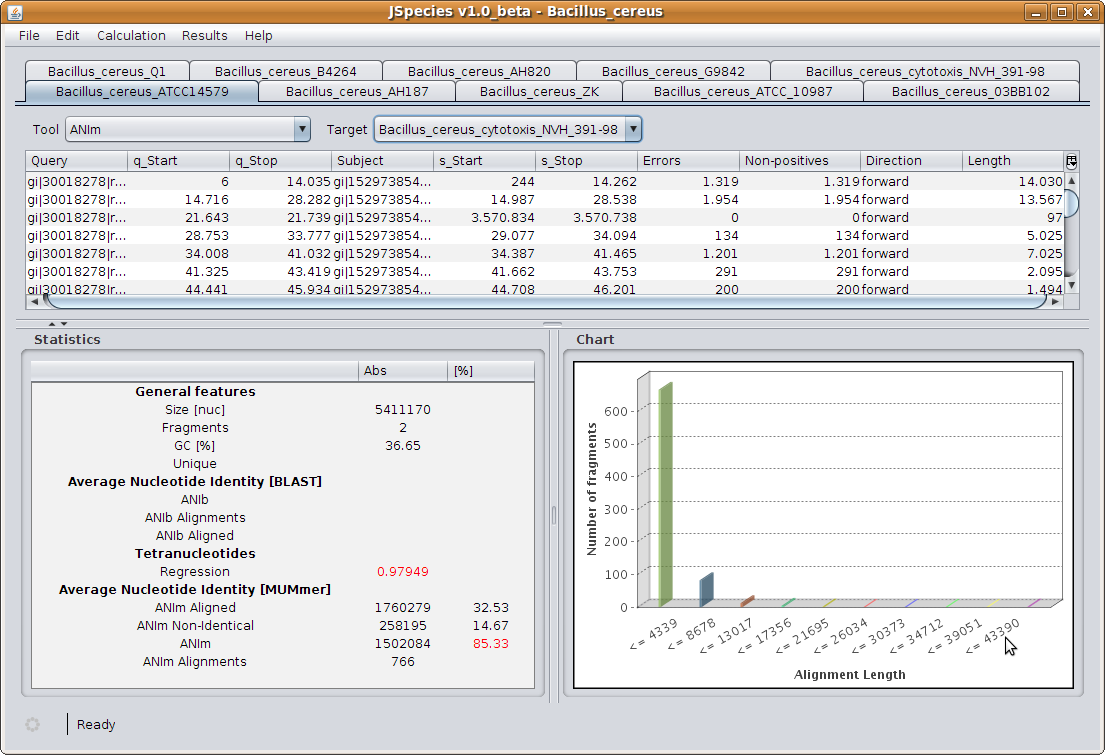
JSpecies is able to calculate the average nucleotide average (ANI) based on BLAST (ANIb) and MUMmer (ANIm).
ANI values are based on pairwise alignment of the genome stretches. The reliability of their results is directly
dependent on the quantity and quality of the aligned DNA fragment. We have evaluated a new parameter based on
oligonucleotide signature frequencies (Tetra) in order to assess whether an alignment-free genomic feature can be
used to circumscribe species. In this regard, we have implemented the use of the tetranucleotide signature frequencies
in our pairwise genome comparisons.
The calculation of ANIb values are implemented as described by:
Average Nucleotide Identity (ANI):
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. (2007) DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 57(Pt 1):81-91
Calculation of tetra nucleotide frequencies and correlation coefficients has been implemented into JSpecies based on the previously described algorithm:
Tetra:
Teeling, H., Meyerdierks, A., Bauer, M., Amann, R. & Glöckner, F.O. (2004)
Application of tetranucleotide frequencies for the assignment of genomic fragments.
Environ Microbiol 6: 938-947
Homepage: www.megx.net/tetra
ANIm values are calculated by using the MUMmer software, in particular the NUCmer (NUCleotide MUMmer) tool:
MUMmer:
Kurtz, S., Phillippy, A., Delcher, A.L., Smoot, M., Shumway, M., Antonescu, C., & Salzberg, S.L. (2004) Versatile and open software for comparing large genomes.
Nucleic Acids Research, Vol. 30, No. 11 2478-2483
Homepage: MUMmer
The Performance
Coming soon.
Interpretation of the Results
More coming soon.
Warning results: for those results that may indicate too low values to be included in the same taxon, the results will be highlighted in red. I.e. values of ANIb and ANIm below 96%, and TETRA values below 0.99
Possible Pitfalls
Coming soon.
License
The current version of JSpecies is released under the GNU General Public License, as published by the Free Software Foundation version 3 of the License.
The JSpecies source code will be available soon in a public repository.
From www.gnu.org:
In the Free Software Movement, we believe computer users should have the freedom to change and redistribute the software that they use. The “free” in free software refers to freedom: it means users have the freedom to run, modify and redistribute the software. Free software contributes to human knowledge, while non-free software does not. Universities should therefore encourage free software for the sake of advancing human knowledge, just as they should encourage scientists and other scholars to publish their work.
System Requirements
- - Linux (recommended) or Windows operating system
- - Sun Java, version 6
- - at least 512Mb of RAM
- - BLAST 2.2.18 (inclusive fastcmd and formatDB)
- - MUMmer 3.0
Information: JSpecies was developed and fully tested under Ubuntu 8.10 / 9.04 with Java 6u13. It is recommended to use at least JavaSE 6u10 in favor for the new Java L&F Nimbus.
Contact
JSpecies has been developed by Michael Richter (mrichter(at)mpi-bremen.de) and Ramon Rosselló-Mora (rossello-mora(at)uib.es ) for the Marine Microbiology Group at the IMEDEA, Esporles, Illes Balears, Spain.
Information: Please send problems and bug reports to one of the developers. We are seeking for Java developers who are willing to take care about bug fixes and enhancements for the Windows / Mac OS version of JSpecies. If you are interested to be involved, please leave us a message.
Google Analytics.
This website uses Google Analytics, a web analytics service provided by Google, Inc. ("Google"). Google Analytics uses "cookies", which are text files placed on your computer, to help the website analyze how users use the site. The information generated by the cookie about your use of the website (including your IP address) will be transmitted to and stored by Google on servers in the United States. Google will use this information for the purpose of evaluating your use of the website, compiling reports on website activity for website operators and providing other services relating to website activity and internet usage. Google may also transfer this information to third parties where required to do so by law, or where such third parties process the information on Google's behalf. Google will not associate your IP address with any other data held by Google. You may refuse the use of cookies by selecting the appropriate settings on your browser, however please note that if you do this you may not be able to use the full functionality of this website. By using this website, you consent to the processing of data about you by Google in the manner and for the purposes set out above.
Citing JSpecies
Richter M, & Rosselló-Móra R (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA
106(45):19126-31.